Thalassemia
Content
• Thalassemia
• Classification
• Pathophysiology
• Clinical features
• Treatment
Objective
At the end of this PDF Note, students will be able to
• Define thalassemia
• Explain the type of thalassemia
• Describe the pathophysiology, clinical signs and symptoms, and treatments for the alpha and beta forms of thalassemia.
Thalassemia
• Thalassemia are a heterogenous group of genetic disorders of Hb synthesis characterized by a lack or decreased synthesis of globin chains
• Two major types of thalassemia:
– Alpha (α) – Caused by defect in rate of synthesis of alpha chains.
– Beta (β) – Caused by defect in rate of synthesis in beta chains.
• Alpha thalassemia usually caused by gene deletion; Beta thalassemia usually caused by mutation.
• Results in microcytic, hypochromic anemias of varying severity
BASICS – 3 Types of Hb
1. Hb A – 2α and 2β chains forming a tetramer
• 97% adult Hb
• Postnatal life Hb A replaces Hb F by 6 months
2. Fetal Hb – 2α and 2γ chains
• 1% of adult Hb
• 70-90% at term. Falls to 25% by 1st month and progressively
3. Hb A2 – Consists of 2 α and 2 δ chains
• 1.5 – 3.0% of adult Hb
INHERITANCE
• Autosomal recessive
• Beta thal – point mutations
on chromosome 11
• Alpha thal – gene deletions on chromosome 16

Classification of thalassemia
• If synthesis of α chain is suppressed – level of all 3 normal Hb A (2α ,2β),A2 (2α,2δ), F(2α ,2γ) reduced
– alpha thalassemia
• If β chain is suppressed- adult Hb is suppressed – beta thalassemia
α-thalassemia
Hb H (β4)
Hb-Bart’s (4)
β-thalassemia
• β+ thal : reduced synthesis of β globin chain, heterozygous
• β 0 thal : absent synthesis of β globin chain, homozygous—— Hb A – absent
Hb F (α22)
Hb A2 (α2 δ2)
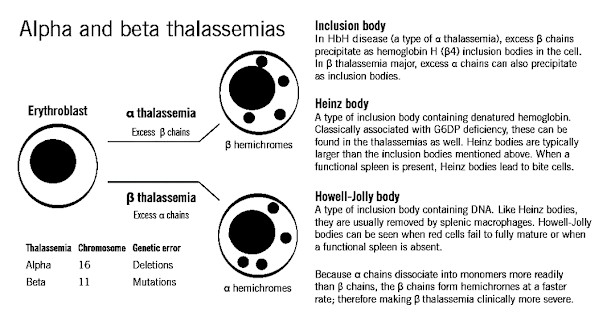
Classification of β Thalassemia
| CLASSIFICATION | GENOTYPE | CLINICAL SEVERITY |
| β thal minor/trait | β/β+, β/β0 | Silent |
| β thal intermedia | β+ /β+, β+/β0 | Moderate |
| β thal major | β0/ β0 | Severe |
Classification OF α-Thalassemia
| No. Of genes present | Genotype | Clinical classification |
| 4 genes | αα/αα | Normal |
| 3 genes | αα/- α | Silent carrier |
| 2 genes | – α/- α or αα/- – | α thalassemia trait |
| 1 gene | –α/- – | Hb H Ds |
| 0 genes | – -/- – | Hb Barts / Hydrops fetalis |
α-Thalassaemia Molecular Pathogenesis
• Defective synthesis of α-globin chains: HbA, HbA2 and HbF
1. Four α-gene deletion: Hb Bart’s hydrops foetalis
2. Three α-gene deletion: HbH disease
3. Two α-gene deletion: α-thalassaemia trait
4. One α-gene deletion: α-thalassaemia trait (carrier)
β–Thalassaemia Molecular Pathogenesis
• β-thalassaemias are caused by decreased rate of β-chain synthesis resulting in reduced formation of HbA in the red cells.
i) Transcription defect
ii) Translation defect
iii) mRNA splicing defect
3 Types of β-Thalassaemia
1) Homozygous form
2) β-Thalassaemia intermedia
3) Heterozygous form
Pathophysiology
• Since ẞ chain synthesis reduced –
• 1. Gamma and delta δ2 chain combines with normally produced α chains ( Hb
F (α22) , Hb A2 (α2
δ2) – Increased production of Hb F and Hb A2
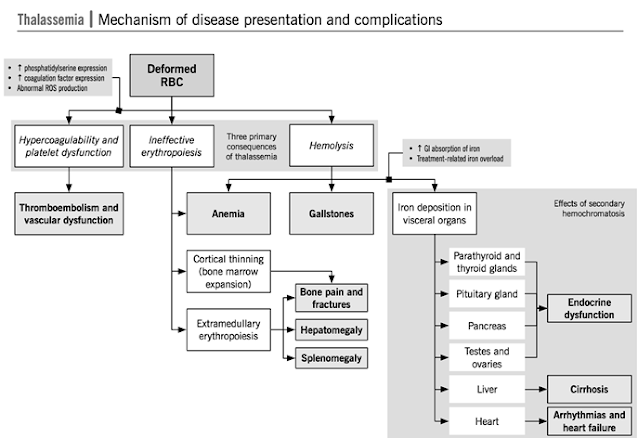
• 2. Relative excess of α chains → α tetramers forms aggregates →precipitate in red cells → inclusion bodies → premature destruction of maturing erythroblasts within the marrow (Ineffective erythropoiesis) or in the periphery (Hemolysis)→ destroyed in spleen
Anemia result from lack of adequate Hb A
→ tissue hypoxia→↑EPO production →
↑ erythropoiesis in the marrow and sometimes extramedullary → expansion of medullary cavity of various bones
Liver spleen enlarge → extramedullay
hematopoiesis
Clinical Features (Thal Major)
INFANTS:
• Age of presentation: 6-9 mo (Hb F replaced by Hb A)
• Progressive pallor and jaundice
• Cardiac failure
• Failure to thrive, gross motor delay
• Feeding problems
• Bouts of fever and diarrhea
• Hepatosplenomegaly
BY CHILDHOOD:
• Growth retardation
• Severe anemia-cardiac dilatation
• Transfusion dependant
• Icterus
• Changes in skeletal system
Clinical Features (Thal Intermedia)
• Moderate pallor, usually maintains Hb >6gm%
• Anemia worsens with pregnancy and infections (erythroid stress)
• Less transfusion dependant
• Skeletal changes present, progressive splenomegaly
• Growth retardation
• Longer survival than Thal major
Clinical Features (Thal Minor)
• Usually ASYMPTOMATIC
• Mild pallor, no jaundice
• No growth retardation, no skeletal abnormalities, no splenomegaly
• MAY PRESENT AS IRON DEFICIENCY ANEMIA (Hypochromic microcytic anemia)
• Unresponsive/ refractory to Fe therapy
• Normal life expectancy
Prevention of Thalassaemia
• antenatal diagnosis
• amniocentesis and foetal DNA studied by PCR amplification technique for presence of genetic mutations of thalassaemias
• Treatment- blood transfusions (4-6 weekly), chelation therapy, folic acid supplement, Bone marrow transplantation
Summary
• Thalassemia are a heterogenous group of genetic disorders of Hb synthesis characterized by a lack or decreased synthesis of globin chains
– Alpha (α) – Caused by defect in rate of synthesis of alpha chains.
– Beta (β) – Caused by defect in rate of synthesis in beta chains.

Also, Visit: Health and Wellness