Applications of Computational tools in Drug Design & Discovery
Importance of CADD
• Traditional drug discovery includes
random screening, serendipitous discovery and process optimization
• Process takes nearly a decade to
complete with an average expense of ~300 million dollar
• CADD tends to curtail this
expenditure and timeline by providing holistic view
• Evolution of CADD began in 1900s
when Emil Fischer (1894) and Paul Ehlrich (1909) propagated the concept of
receptors and lock and key mechanisms
• Scientific advancements during the
past two decades have changed the way pharmaceutical research generate novel
bioactive molecules
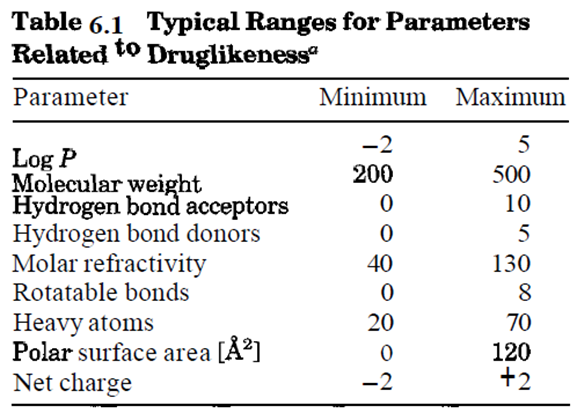
Druglikeness Screening
• Many
drug candidates fail in clinical trials because of reasons unrelated to potency
against the intended drug target
• Pharmacokinetics
and toxicity issues are blamed for more than half of all failures in clinical
trials
• First
part of virtual screening evaluates the druglikeness of small molecules
• Druglike
molecules exhibit favorable absorption, distribution, metabolism, excretion,
and toxicological (ADMET) parameters
Discovery of ACE Inhibitor- Captopril
• Angiotensin converting enzyme is a
carboxypeptidase with a zinc ion as cofactor
• Plays a key role in the
renin-angiotensin cascade involving blood pressure control
• Captopril, a clinically important,
potent and reversible inhibitor of ACE
• Design of captopril is one the early
endeavours and success of structure based drug design
Information
collected for SBDD
• Enzymatic mechanism of ACE was
similar to that of carboxypeptidase A
• Exceptional is ACE cleaves off a
dipeptide whereas a carboxypeptidase A cleave a single amino acid residue from
carboxyl end of a protein
• L-benzylsuccinic acid is a potent
inhibitor of carboxypeptidase A
• BPP5a (HOOC-Glu-Lys-Trp-Ala-Pro-NH),
a potent pentapeptide inhibitor of ACE that was isolated from the venom of the
Brazilian viper
• Captopril is the first ACE inhibitor
to enter clinical use following its approval by USFDA in 1981
• Frontline therapeutic agent for the
treatment of hypertension and heart
failure

Proline

Selectivity or promiscuity? Or both?
• Modern drug development projects
should aim to deliver target specific active compounds
• Approach should be- one disease, one
target and one drug
• Retrospective analysis proved that
approved drugs are promiscuous and bind to several target proteins
• Property of active compound binding
to multiple proteins is termed as polypharmacology
• Sorafenib- A Raf inhibitor
originally developed against lung or pancreatic cancer
• Proved effective against renal cell
cancer by its action on VEGFR2 receptors
• Paxil- A serotonin uptake inhibitor
also binds to beta adrenergic receptors offering plausible explanation for
increased heart rate
• Prediction of compound
polypharmacology has the potential to identify possible adverse effects
• Several methods have been developed
for computational prediction of compound polypharmacology
• At present over 5000 drugs, ~10
million virtual library compounds and 1,45,219 biological macromolecule
structures are available for exploration which are publicly accessible
• Efficient polyphamacology prediction
may be helpful in the future to facilitate drug repurposing and
• To discover more potent drug with
less off-target toxicity
Structure of protein
• Have to focus on detailed three
dimensional structure of biological molecules
• Like shape or structure of a protein
offers clues about the role it plays in the body
• Proteins are shaped to get their job
done
• May help in developing new medicines
or diagnostic
• Design of lock helps in making
key
Proteins are the body’s worker molecules

X-ray Crystallography

Strategies for drug design
• Molecular docking and Dynamics
• Quantitative Structure Activity
Relationship
• Pharmacophore modelling
Virtual Screening
• Assessment
of overall drug likeness
• Ability
to specifically bind to a given drug target
• Goal-
reduction of enormous virtual chemical space of small organic molecules to
synthesize and/or screen against a specific target to a manageable number of
compounds that exhibit the highest chance to lead to drug candidate
• Source
of information
• What
does a drug look like in general?
• What
is known about compounds that interact with the receptor?
• What
is known about the structure of target protein and protein-ligand interactions?
• Virtual
screening is a category of in silico methods that can be utilized to identify
molecules that will (potentially) bind to a target of interest
• These
methods are classified as either structure-based or ligand-based:

Before commencing a screen, ask yourself “given the data I
have, which virtual screening tool is most appropriate?”
LBDD
• Ligand-Based Drug Design (or
indirect drug design):
• Relies on knowledge of other
molecules that bind to the biological target of interest
• May be used to derive a
pharmacophore model that will define the minimum necessary structural characteristics
a molecule must possess in order to bind to the target
SBDD
• Structure-Based Drug Design (or
direct drug design):
• Relies on knowledge of the three
dimensional structure of the biological target
• Obtained through methods such as
X-ray crystallography or NMR spectroscopy
• Using the structure of the
biological target, candidate drugs are predicted that will bind with high
affinity and selectivity to the target
• Interactive graphics and the
intuition of a medicinal chemist are further used in this design process
SP & XP modes of docking

• High Throughput virtual screening
(HTVS)
• SP- Standard precision and XP- Extra
Precision
• XP scoring function include more
stringent terms like hydrophobic effects and charged interactions
• Induced-fit docking (IFD)
• Molecular Dynamics
Chemical compound repositories
|
Database |
Sample |
|
PubChem |
~40,000,000 |
|
Accerlrys |
~7,000,000 |
|
PDBeChem |
~14,572 |
|
Zinc |
~21,000,000 |
|
LIGAND |
~16,838 |
|
DrugBank |
~6711 |
|
ChemDB |
~5,000,000 |
|
WOMBAT |
~331,872 |
|
MDDR |
~180,000 |
|
3D MIND |
~100,000 |

Quantitative Structure Activity Relationship
• Most popular approach
• QSAR- computational method to
quantify the correlation between chemical structures of series of compounds and
a particular chemical or biological process
• Hypothesis behind the concept is
similar structural or physicochemical properties have similar activity
Methodology of QSAR
• Identification of ligands with
experimentally measured values of desired biological activity.
• Should be of adequate chemically
diversity to have large deviation in activity
• Identify and determine molecular
descriptors associated with various structural and physico-chemical properties
of the molecules under study
Definition of
molecular descriptor
• The
molecular descriptor is the final result of a logic and mathematical
procedure which transforms chemical information encoded within a symbolic
representation of a molecule into a useful number, or the result of some standardized
experiment
• Roberto Todeschini and Viviana Consonni
What are descriptors?
Includes
molecular weight,
Lipophilicity
Hydrogen
bonding donors & acceptors
Molecular
connectivity
Molecular
topology
Molecular
geometry
Stereochemistry
Good descriptors
should characterize molecular properties important for molecular interactions
Literature
suggests that more than 2000 molecular descriptors can be calculated

• Discover correlations between
molecular descriptors and the biological activity that can explain the
variation in activity in the data set
• Test the statistical stability and
predictive power of the QSAR model
• Goal here is to create a molecular
“fingerprint” for each molecule that relates to its activity
• Essential part of the drug optimization
process
• Success of any QSAR model greatly depends on the
a) choice of molecular descriptors and
b) ability to generate the appropriate
mathematical relationship between the descriptors and the biological activity
of interest
• Statistical methods applied in QSAR:
a)
Multivariable linear regression analysis (MLR)
• Simplest method to quantify the
molecular descriptor having good correlation with the variation in activity
• For large numbers of descriptors the
MLR method can be time consuming
b) Principle
Component Analysis (PCA)
• Efficient method for reduction of
the number of independent variables
• Highly useful for systems with a
larger number of molecular descriptors than the number of observations
• Results from PCA are often difficult
to analyze
c) Partial
Least Square analysis (PLS)
• Combination of MLR and PCA
techniques
• Gives good correlation
• Advantageous for systems with more
than one dependent variable
• Biological systems often display
non-linear relationship between the molecular descriptors and the activity
• Once an initial QSAR model has been
developed is must be validated
• By internal validation and external
validation
Manikanta et
al., European Journal of Medicinal Chemistry, 2017, 130, 154-170
Pharmacophore modelling
• Describe 3D features of a molecule
• Molecular descriptors are then
combined to create a pharmacophore that can explain the biological activity of
the ligands
• A pharmacophore is defined as a
spatial arrangement of functional groups and substructures common to active
molecules and essential to biological activities
• On the concept of “molecular
similarity” of small molecules are derived from a series of active compounds
and inactive ones
• Pharmacophore- qualitative aspect
Pharmacophore
• Describe the two or three
dimensional arrangement of physicochemical properties of a compound.
• Can be used to screen for molecules
with similar arrangement of features.
• In phase , the properties
Ø
Include A (acceptor), D (donor), H
(hydrophobic), N (Negative), P (positive), and R (aromatic).
Ø
Have defined geometry (point, vector or group).
Ø
Are defined via SMARTS patterns.

How to create a
hypothesis manually
1. Prepare molecule:
·
Prepare Ligands (LipPrep)
·
Generate Conformers (CobfGen)
2. Create a common hypothesis:
·
Align known actives and identify common features
3. Screen a library:
·
Identity compounds that match all or most
features
·
Tweak hypothesis if necessary
Shape based screening
• Hard-sphere overlaps
• Conformers for a screening structure
B are aligned to a template structure A.
• Hundreds of trial alignment are
considered for each conformer of B.
• The
conformer and alignment with the largest A-B overlaps wins.

Conclusion
• CADD- moving drugs from concept to
the clinic
• Computational structure-based design
supported by medicinal chemistry strategies, can lead to the development of
drugs or
• Drug-like molecules with refined
pharmacological activity that is better than the parent molecule
• SBDD has been recognized as the tool
that facilitated the development of several important drugs in current clinical
use or late stage clinical development
• Allowing many drug discovery
scientists to carry out more focused, hypothesis-driven discovery initiatives
limiting the number of compounds that are synthesized
• Adoption of early stage PK and PD
studies has also contributed greatly to the significantly reduced late-stage
attrition rate of clinical candidates