Urea Cycle

Objective
• At the end of this lecture, student will be able to
– Explain urea cycle
– Explain significance of urea cycle
Overview of amino acid catabolism in mammals

UREA CYCLE
• Urea cycle / Krebs-Henseleit cycle
• Urea is the end product of protein metabolism (amino acid metabolism)
• Nitrogen of amino acids is converted to ammonia
• Ammonia is toxic to the body and hence converted to urea and detoxified
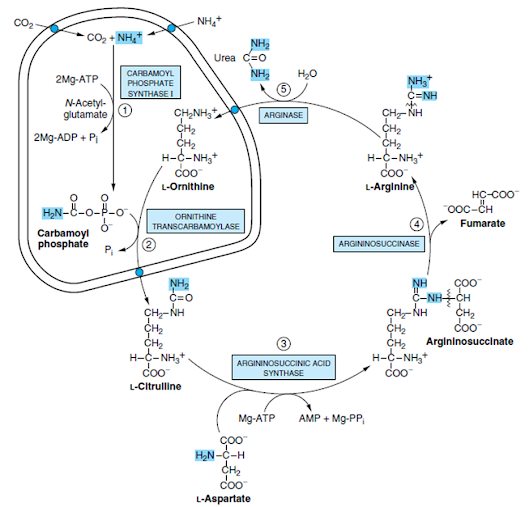
• Urea synthesis is a five-step cyclic process, with five distinct enzymes
• First two enzymes are present in mitochondria while the rest are located in cytosol
• Urea is synthesized in liver and transported to kidneys for excretion in urine
• Urea cycle was elucidated by Hans Krebs and Kurt Henseleit (1932), hence it is known as Krebs-Henseleit cycle
• Urea has two amino (-NH2) groups, one derived from NH3 and the other from aspartate & carbon atom is supplied by CO2

• Interrelation between urea cycle and TCA cycle

Step I: Synthesis of carbamoyl phosphate
• Carbamoyl phosphate synthase I (CPS-l) catalyses the condensation of NH3 ions with CO2 to form carbamoyl phosphate
• This step consumes 2 ATP and is irreversible and rate-limiting
• CPS I requires N-acetylglutamate for its activity
• Takes place in mitochondria
• CPS-I is rate-limiting enzyme of the urea cycle, is active only in the presence of its allosteric activator N-acetyl glutamate, which enhances the affinity of the synthase for ATP
Step II. Formation of citrulline
• Citrulline is synthesized from carbamoyl phosphate and ornithine by ornithine transcarbamoylase
• Ornithine is regenerated and used in urea cycle
• Therefore, its role is comparable to that of oxaloacetate in citric acid cycle
• Ornithine and citrulline are basic amino acids
• Citrulline produced in this reaction is transported to cytosol by a transporter system
Step III. Synthesis of arginosuccinate
• Arginosuccinate synthase condenses citrulline with aspartate to produce arginosuccinate.
• The second amino group of urea is incorporated in this reaction.
• This step requires ATP which is cleaved to AMP and pyrophosphate (PPi).
• The latter is immediately broken down to inorganic phosphate (Pi)
Step IV. Cleavage of arginosuccinate
• Arginosuccinase cleaves arginosuccinate to give arginine and fumarate
• Arginine is the immediate precursor for urea
• Fumarate liberated here provides a connecting link with TCA cycle, gluconeogenesis etc
Step V. Formation of urea
• Arginase is the fifth and final enzyme that cleaves arginine to yield urea and ornithine
• Ornithine, so regenerated, enters mitochondria for its reuse in the urea cycle
• Liver can ultimately produce urea
Overall reaction and energetics
• The urea cycle is irreversible and consumes 4 ATP
• 2 ATP are utilized for the synthesis of carbamoyl phosphate
• 1 ATP is converted to AMP and PPi to produce arginosuccinate which equals to 2 ATP
• Hence 4 ATP are actually consumed
NH4+ + CO2 + Aspartate + 3ATP —–→ Urea + Fumarate+ 2 ADP + 2 Pi + AMP + PPi
Regulation of urea cycle
• The first reaction catalysed by CPS-l is rate limiting reaction in urea synthesis
• CPS-I is allosterically activated by N-acetylglutamate and degraded by a hydrolase
• The rate of urea synthesis in liver is correlated with the concentration of N-acetylglutamate
• High concentrations of arginine increase NAG, increases the level of NAG in liver, leading to enhanced urea synthesis
Disposal of urea
• Urea produced in the liver freely diffuses and is transported in blood to kidneys, and excreted
• A small amount of urea enters the intestine where it is broken down to CO2 and NH3 by the bacterial enzyme urease, this ammonia is either lost in the faeces or absorbed into the blood.
• ln renal failure, the blood urea level is elevated (uremia), resulting in diffusion of more urea into intestine
• Hyperammonemia (increased blood NH3) is commonly seen in patients of kidney failure
• Major changes in diet can increase the concentrations of individual urea cycle enzymes 10-fold to 20-fold
• Starvation, for example, elevates enzyme levels, presumably to cope with the increased production of ammonia that accompanies enhanced protein degradation
Metabolic disorders are associated with each reaction of the urea cycle
• All the disorders invariably lead to hyperammonemia and toxicity
• The clinical symptoms associated with defect in urea cycle enzymes Include vomiting, lethargy, irritability, ataxia and mental retardation
(1) Defects in any of several enzymes of a metabolic pathway enzyme can result in similar clinical signs and symptoms
(2) The identification of intermediates and of ancillary products that accumulate prior to a metabolic block provides insight into the reaction that is impaired
(3) Precise diagnosis requires quantitative assay of the activity of the enzyme thought to be defective
(4) Rational therapy must be based on an understanding of the underlying biochemical reactions in normal and impaired individuals

Summary
• Urea cycle is the end product of protein metabolism
• Urea is synthesized in liver and transport to kidney for excretion in urine
• 4 ATP are consumed in urea cycle
• Normal blood urea concentration is 10 – 40 mg/dl
• Hyperammonemia is increase in ammonia level in blood
• CPS-l is rate limiting reaction in urea synthesis
